从AlphaFold到RoseTTAFold:一位生物信息学工程师亲历蛋白质结构预测革命
2020年盛夏,当我第一次在arXiv上看到AlphaFold2的论文时,整个人都愣住了。作为一名在生物信息领域摸爬滚打近十年的老兵,我太清楚蛋白质结构预测这道难题的分量——它被称为“21世纪生物学的圣杯”,困扰了科学家整整五十年。
而DeepMind用注意力机制把这杯酒一饮而尽。
转折时刻:AI打开了潘多拉魔盒
那一年的冬天,我几乎把所有业余时间都泡在了AlphaFold2的开源代码里。反复调试、反复测试,我发现了一个核心秘密:Transformer架构之所以能hold住蛋白质这种超长序列,靠的是多头注意力机制对氨基酸残基间关系的全局建模。简单说,AI学会了“隔空对话”——即使两个氨基酸在序列上相隔甚远,也能捕捉到它们在三维空间里可能产生的相互作用。
但AlphaFold2有一个软肋:速度。
一次完整预测,动辄需要数小时甚至数天。这对于需要批量处理的研究场景而言,简直是噩梦。
破局者:RoseTTAFold的三轨智慧
2021年盛夏,DavidBaker团队给出了答案。我在第一时间研读了那篇NatureMethods论文,心得是:RoseTTAFold的设计哲学与AlphaFold2截然不同。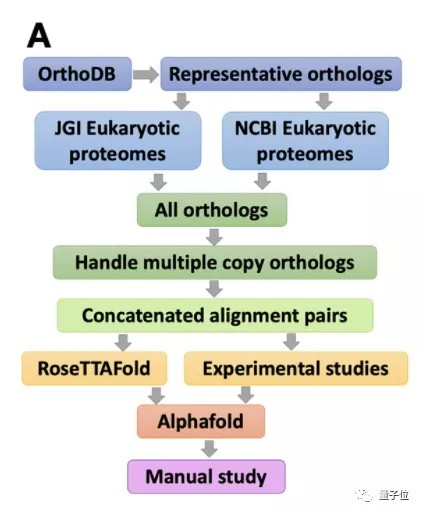
它采用三轨注意力机制,同时追踪蛋白质的一级结构(氨基酸序列)、二级结构(α螺旋、β折叠)、以及三级结构(完整3D折叠)。三个维度之间设置多重连接,神经网络能够端到端学习三个层次的联合分布。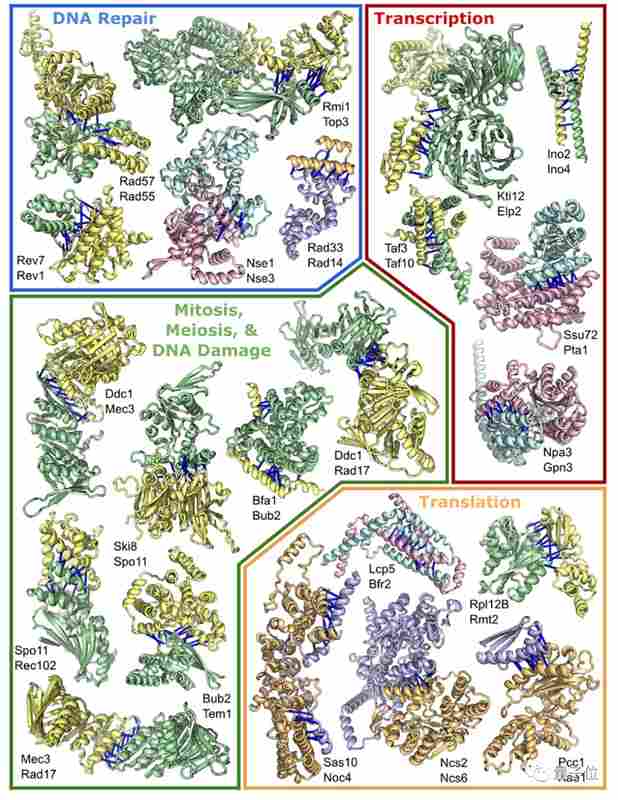
关键在于那个双轨道变体模型:牺牲少量准确率,换取100倍的推理速度提升。这是一笔精妙的交易——对于大规模筛选场景,“够快且够准”远比“极致精准但慢如蜗牛”更有实用价值。
双剑合璧:复合体预测的正确姿势
真正让我拍案叫绝的,是最新Science论文展示的组合策略。
研究团队的核心思路非常清晰:先用协同进化分析在酵母菌基因组中捞出830万对蛋白质,筛选出1505种可能形成复合体的候选者;再让RoseTTAFold和AlphaFold2分别预测这些候选者的三维结构;最后通过交叉验证确定最终的模型。
结果:700多种此前结构未知的蛋白质复合体获得了可靠的3D预测,更有106种属于全新发现。
我反复咀嚼这个数字背后的意义。106种“从未被描述过的全新蛋白质复合体”——这意味着自然界里还有大量未知的生命机器等待我们去发现,而AI就是那把钥匙。
工程实践:如何复用这套方法
对于想复现这套流程的同行,我有几点建议。
第一,协同进化分析是基础。推荐使用JackHMMER或HHblits构建多序列比对(MSA),确保覆盖足够广泛的物种多样性,否则后续的预测就是无源之水。
第二,合理分配计算资源。用RoseTTAFold做初筛、AlphaFold2做精修,这种“粗筛+精调”的二级架构能让你在有限GPU资源下最大化产出。
第三,不要迷信自动预测。对于关键靶点,建议结合人工检查——特别是那些预测置信度处于边缘区间的模型,很可能隐藏着假阳性或假阴性。
结构生物学的游戏规则已经改变。计算将扮演根本性角色——这是论文里的原话,也是我这几年最深刻的体会。拥抱它,适应它,你就能站在浪潮之巅。



